Introduction
이 글은 원자재 및 완제품의 테스트에 대한 언급도 있지만 주로 박테리아와 바이러스 백신을 포함한 생물학적 제제에 적용되어야 하는 환경 모니터링(EM, Environmental Monitoring) 프로그램에 관한 것이다. 생물학적제제의 산물은 세포 배양 및 발효, 적용 물질의 추출, 정제를 통해 세포, 조직 또는 미생물에서 파생된다. 제품은 항원, 호르몬, 효소, 항체, 면역 혈청, 발효 제품, 박테리아 및 바이러스 백신을 포함한다.
이 글의 많은 부분은 일반적으로 무균의약품의 제조 및 충전 시 환경 모니터링에 적용될 것이다. 일부 백신은 여과할 수 없거나 열, 가스 또는 방사선에 의해 멸균될 수 없기 때문에 무균 처리는 주로 무균성 보장을 위해 사용된다. 충전기 등 설비의 무균 처리뿐만 아니라 환경 관리 및 모니터링, 탱크 및 기타 장비의 청소 및 소독, 가능한 경우 Closing system 및 SUS(Single Use System)을 사용해야 한다.
따라서 제품 오염이나 교차 오염 관리를 보장하고 허용 가능한 환경 제어 수준을 보장하기 위해 생물 안전 시스템(Bio safety system)과 cGMP 시스템 간에 긴밀한 협업이 이루어져야 한다. 이는 특히 백신 준비를 위해 살아있는 유기체 또는 포자를 형성하는 유기체, 특히 생물 안전 수준 (BSL, Bio safety Level)3과 4(음압 영역)은 유기체의 성장에 적용된다. 세계보건기구(WHO)의 문서에 따르면 Seed lot와 Cell bank를 구축하는 동안 같은 지역에서 다른 전염성 물질이나 살아있는 물질과 함께 작업해서는 안 된다. 항원성을 유지하면서 활성 또는 감쇠된 백신과 다른 비활성 수단(화학물질, 열 등)을 위한 전용 시설의 사용을 고려해야 한다.

Environmental Control Measures
환경 관리의 방법으로는 차압(differential pressures), 환기횟수(air changes), 단일 방향 공기 흐름(unidirectional air), 가능한 한 Closing 된 제조 공정, gowning 등의 Qualified 된 공정, Media fill test, Bioburden의 모니터링 등이 있다. HEPA Filter는 6개월마다 qualified 되어야 하며, 3년마다 가장 높은 관리 구역(Grade A)에 대해 Smoke test를 수행해야 한다. 샘플 절차, 샘플링 유형 및 빈도, 경고 및 조치 수준 및 경향을 정의 해야 하며, 레벨이 초과되거나 경향이 감지될 경우 조사를 실시해야 한다.
원자재 및 작업원의 동선은 Grade D 구역에서 시작하여 Grade C로, Grade B로, 마지막으로 Grade A로 흐름을 잘 정의해야 한다. Grade C와 Grade D에 대한 특정 PPE(Personal Protective Equipment)는 마스크와 장갑 그리고 일체형 작업복과 같은 것을 포함한다. 한 번에 APA(Aseptic Processing Area)에서 허용되는 작업원 수, APA(Aseptic Processing Area)에 체류할 수 있는 기간 및 개입 관리(inherent/routine and corrective)에 제한이 있어야 한다. 작업원이 APA(Aseptic Processing Area)에 들어오기 전에 적격성을 Qualified 해야 하며, Media fill test 중에 적절한 무균 작업 기술이 확인 되어야 한다.
공기의 Bioburden, 작업원들, 용수와 가스가 관리 되어야 한다. 샘플은 샘플링의 주기 및 절차, 종류는 표준 작업 절차서에(SOPs)규정되어야 한다. 관리 SOPs에 관리 한계나 경향이 확인되어 이를 초과하면 취할 행동에 대하여 규정되어야 한다.
소독제는 시설에 존재하는 표면에 대하여 시험을 통해 Qualified 되어야 한다. 승인된 소독제, 희석 및 적절한 접촉 시간은 SOP에 명시되어야 한다.
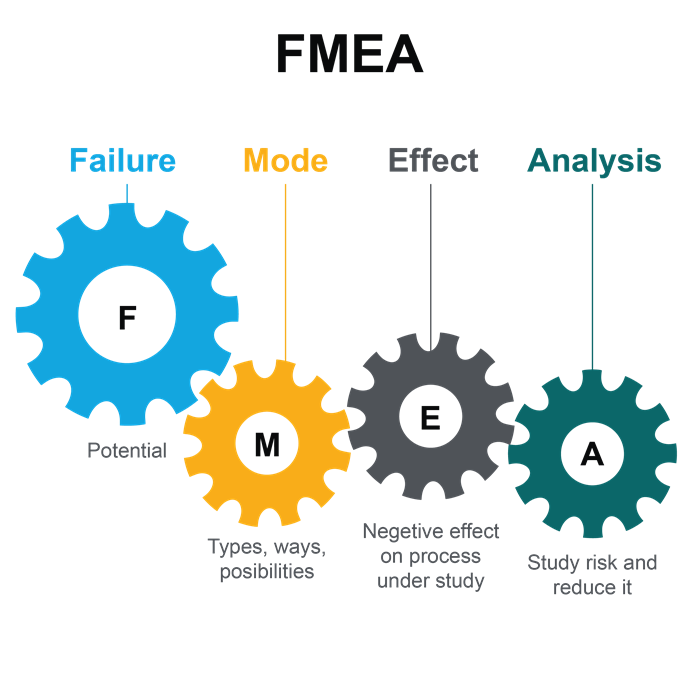
Failure Modes and Effects Analysis
미생물 오염원을 평가하기 위한 FMEA(Failure Mode and Effect Analysis)는 무균 처리 영역으로의 유기체의 침투를 막기 위해 어떤 조치 또는 변경해야 하는지 결정하기 위한 귀중한 문서다. 위험 기준은 앞에 정의되어야 한다(점수(scoring) 및 허용 가능성(acceptability) / 임계값 수준(threshold levels)). 위험 우선순위 번호(RPN, Priority Number)는 다음과 같이 계산된다. Severity x Probability of Occurrence x Detection = RPN
임계값(threshold levels)을 사용하여 확인된 위험(높은 위험, 중간 위험, 낮은 위험)의 중요성을 파악한다.
완화 평가 결과(즉, 높은 위험, 높은 우선순위)에 따라 리스크의 우선순위를 정한다.
완화 결정 및 활동은 멸균성 보증, 품질 보증, 품질 관리, 규제 준수, 생산, 엔지니어링 및 검증으로 구성된 SME(Subject Matter Expert) 팀에 의해 결정되어야 한다.
Testing of Raw Materials, Bulk and Finished Product
원자재, 혈청 및 배지를 시작으로 테스트를 수행하고 전염성해면양뇌증(TSE, Transmissible Spongiform Encephalopathy)이 없음을 보장한다. Bioburden은 도움이 되지만 꼭 필요한 테스트는 아니다. 제품 무균성 테스트 완료, 외부 바이러스 부재, Mycoplasma 부재를 보여주는 외래성 물질 테스트가 수행되어야 한다.
" EM of Clean Rooms in Vaccine Manufacturing Facilities" (WHO 2012)에 따르면, 백신 생산의 다양한 단계에 대한 클린룸 등급을 결정하기 위해 위험 기반 접근법을 사용한다. 원자재는 Grade A 또는 최대 Grade B 또는 Grade A의 Isolator(Back ground: Grade D)에서 샘플링 해야 한다. 최종 제품이 여과 멸균될 경우 샘플링이나 기타 처리 단계를 위해 최소한 Grade C에서 수행 해야 한다.
백신이 병원성 유기체인 경우 환경과 사람으로부터 제품을 보호하고 병원 감염으로부터 작업원을 보호하기 위해 단방향 기류(unidirectional airflow) Grade A나 Barrier system을 사용하여 교차 오염을 방지해야 한다. 이 경우 전용 시설의 사용을 고려하거나 제품에 약독화 백신과 항생제 또는 방부제를 만드는 것을 고려 해야 한다. 약독화 백신은 wild type(예: 오래된 단종 경구용 소아마비 백신)으로 환원하기 위한 테스트를 거쳐야 한다.
환경 모니터링은 제형 및 충전 구역의 오염 관리를 표시해야 하며, 이를 통해 오염으로부터 배치를 보호 할 수 있다. 환경 모니터링 결과는 영역의 미생물 제어를 반영한 것임으로 환경 모니터링(EM) 수준과 배치 무균성 사이에는 직접적인 상관관계가 없다. 단, 배치 영향은 환경 모니터링 및 작업원 모니터링(PM) 결과에 기초하여 평가해야 한다. 위험 평가는 해당 평가를 문서화하기 위해 작성될 수 있다.
Cleanroom Standards
클린룸 표준은 FDA(2004년 Aseptic Guidance document), USP <1116> 및 EU GMP Annex 1과 WHO(Annex 4와 Annex 6)에 의해 정의되었으며, 공기 중(Settle plates) 및 표면(RODAC)에 대한 non-viable particle limits (NVP) 와 microbial levels에 관한 것이다. 최근 USP에서는 15 CFU의 특이성이 발견 될 경우 비정상적인 것으로 간주되지만 수치 수준에서 Sample type 및 클린룸, Grade 당 월별 권장 회수율로 전환 되었다.
Non-Viable Particle (NVP) Monitoring
NVP(Non-Viable Particle) 모니터링의 경우 미국(FDA Aseptic Guidance and USP)은 5.0 μm 크기의 입자는 모니터링 하고 있지 않고, 0.5μm 크기의 입자만 모니터링을 요구하지만 EU GMP Annex 1은 더 큰 입자를 모니터링 하도록 요구한다. 그러나 이 요건은 Annex 1 최근 제안된 Update에서 제거되었다. 복수의 작업장에서의 NVP 값을 평균화하지 않는 것이 중요하다. 프로브 위치는 가능한 제품 오염 평가를 위해 개방된 제품(중요 충전 구역에서 약 30 cm)과 최악의 경우 위치에 가까워야 한다. NVP 모니터링 위치를 결정하려면 ISO 14644를 가이드로 사용하는 것이 바람직하다. 또한, 샘플링 위치가 있는 방의 도면이 관리되어야 한다.
Grade A 지역은 전체 작업 활동에 대한 기간의 모니터링을 위하여 고정 NVP 모니터링 카운터를 사용하여 지속적인 모니터링을 수행한다. Portable counters는 추가 위치에 배치할 수 있으며 필요할 경우 연속 모니터링을 교체하는 데 사용할 수 있다. 포자형성 유기체 또는 더 높은 등급의 생물안전시설과 같은 분리된 영역에서는 전용 입자 계수기를 사용해야 한다.
시설은 최소한 연간마다 Grade 별 관리 기준을 충족하고, 새로운 구역의 작업실 분류를 검증할 수 있는 자격을 갖추어야 한다. Grade A와 Grade B의 경우 필터 무결성 시험(Filter integrity Testing)을 6개월마다 수행해야 하며, 이를 실패할 경우 조사와 재확인을 필요로 한다.
Viable Monitoring
Viable monitoring은 환경에 대한 미생물 관리를 보장하며 무균 제품, 백신 및 의약품에 사용된다. 공기의 샘플링은 일정 시간 내에 특정 공기량(1m3)을 포집하는 것이 가장 정량적인 방법이다. Settle plates는 반정량형이며 4시간 동안 노출된다. 대부분의 제약 공장들은 WHO의 요구사항에 따라 전체 운영 기간 동안 또는 교대 시간을 USP <1116>에 다라 커버하기 위하여 두 번 Test를 실시 한다. RODAC은 표면의 세척 효능을 확인하는데 사용된다. (미디어 플레이트에 사용되는 소독제에 대한 올바른 중화제가 있는지 확인) 작업이 끝날 때 장비의 소독제와 무균 작업 구역의 작업원을 샘플링 하기 위한 올바른 절차가 있어야 하며, 손가락, 팔 및 가슴에 대한 일상적인 모니터링을 수행해야 하며 손가락 모니터링은 장갑을 교체하기 전에 다른 충전실로 들어가기 전에 그리고 소독제로 소독하기 전에 행해져야 한다.
Grade A 구역에서는 4시간마다 공기에 대한 모니터링을 실시해야 하며, Settle plates는 해당 기간 내내 열어 두어야 하며, Settle plates은 작업 후(또는 정적 모니터링 중) 및 외부 장갑을 교체하기 전에 샘플링 해야 한다.
환경 모니터링 프로세스에 대한 계획은 신규 시설의 시공 완료 후에 문서화 되어야 하며, 이전 시설은 제품 노출, 최고 생산량 및 청소하기 가장 어려운 지역의 위험 평가를 통해 선정되어야 한다. 특히 (조치 수준이 지침 문서에 정의되어 있기 때문에) 경보 수준을 결정할 때는 모든 위치에 대해 100개 이상의 데이터를 수집하는 것이 가장 좋다. 경고 수준은 경향를 결정하기 위해 사용되어야 하며, 많은 데이터가 축적된 후 통계 분석에 기초하여 수정할 수 있다.
At rest 상태의 모니터링은 소독제 효능에 대한 정보를 제공하며, 생산 작업자가 시설에 들어가기 전에 매월, 분기별로 또는 반년마다 가동 중지 후 수행되어야 하며, 이를 SOP에 정의 해야 한다. In operation은 환경 관리에 관한 정보를 제공한다. 배지 성능 시험 및 소독제 유효성 검사에 사용할 환경 및 작업원의 데이터베이스를 구축해야 한다.
Investigations
경고 수준을 벗어난 부분에 대하여 조사할 필요는 없지만, 이것은 반드시 SOP에 정의되어야 한다. 경고 수준은 항상 조치 수준보다 낮다. 조치 수준은 규제 지침 문서(예: USP, Annex 1, PDA Technical report 13)에 정의되어 있으며 Grade, Clean Room 또는 Sample type 별로 초과할 경우 조사가 필요하다. 경향은 Grade나 Clean room, 미생물 유형별 조사에도 포함될 수 있다. 경향(추세)은 일반적으로 지정된 기간 동안 3개 이상의 연속된 점으로 정의된다. 경향(추세)은 일반적으로 계수의 증가, 반복적인 미생물 회복의 변화 또는 같은 작업실에서 여러 계수의 변화를 보일 경우 역행으로 간주된다.
조사는 Fishbone Method, 5 Whys, Kepner-Tragoe Analytical Trouble-shooting을 포함한 다양한 근본 원인 분석 기법을 사용하여 조사를 수행해야 하지만, 1 CFU에 대한 근본 원인은 발견되지 않을 것이다. 위험평가는 유용한 도구 이다. 미생물 유형의 검토(Gram-negative 미생물은 제품에 잔류 및 endotoxin을 남길 수 있으며, mold는 allergens을 남길 수 있다.) 살균 방법 및 소독의 위치 등을 포함하여 제품에 미치는 영향을 결정하고 CAPA가 Open 되어야 한다. 근본 원인이 발견되지 않았더라도 배치가 오염되었을 가능성이 있는 경우, 해당 배치의 출하 금지 등을 고려 하여야 한다.
발생 가능성을 줄이거나 검출 가능성을 높이는 조정은 합리적인 후속 조치로 간주한다. 높은 위험 활동(위험 평가가 작성된 경우)은 Corrective action 및 Preventive actions으로 이어져야 하며, 궁극적으로 CAPA 유효성에 대하여 평가되어야 한다.

Media Fills
충전 시간, 검증된 intervention, 최대 작업원 등과 같은 제조 공정을 확인 하기 위하여 반년마다 Media fill test가필요 하다. 해당 Test가 실패 할 경우 마지막 성공 이후 충전된 모든 Lot에 대하여 의문을 가지고 조사를 실시해야 하며, 이 후 세 번의 추가 Test가 수행 되어야 한다.
의약품에 대해서는, 인간이 무균 공정에서 가장 큰 오염원이기 때문에 백신 제품 충전을 위한 최고의 시스템은 closed systems, RABs, Isolators 등, 가까운 인간과의 접촉을 최소화 하는 것이 중요 하다.
'My Job > Technical for Pharmaceutical' 카테고리의 다른 글
| 수동 이물검사자 자격의 중요성 (Qualification of Manual Visual inspector) (0) | 2022.10.08 |
|---|---|
| 제조 생산성에 대한 가능성을 보여주는 새로운 Vial 기술 (0) | 2022.10.08 |
| 제약산업의 효과적인 교육 프로그램의 구축 (0) | 2022.10.01 |
| How close are we to closed processing? (1) | 2022.10.01 |
| 염료 침투 테스트(Dye(Blue)Ingress Test)는 적절한가? (1) | 2022.10.01 |