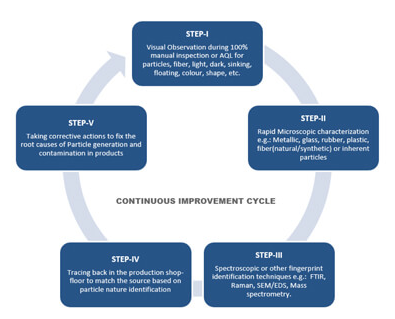
주성분(API, active pharmaceutical ingredients), 부형제, 포장재, 중간체 및 완제품 테스트는 제약 산업 품질 관리 부서의 주요 업무 중 하나이며, 이는 Batch(Lot)의 일부분을 샘플링하여 분석한다. 그런 다음 분석 결과를 사용하여 전체 Batch(Lot)의 품질에 대한 결론을 도출하게 됨으로 올바른 샘플링은 이 과정에서 중요한 역할을 하며 어떤 분석 방법도 아무리 좋은 방법이라도 샘플링 과정에서 발생한 오류를 보상할 수는 없다. Regulatory Requirements for Sampling(샘플링에 대한 규제 요건) 우선 국가별 요구 사항을 고려해야한다. 예를 들어, 독일에서는 ‘의약품 및 주성분 제조에 관한 법령 (Arzneimittel-und Wirkstoff..