A SMART Approach To CAPA Effectiveness Checks
시정 예방 조치(CAPA, Corrective and preventive action) 문제는 FDA에 의한 Form 483 결과의 상위 중 하나이다. 종종, CAPA는 프로세스의 구조와 흐름으로 인해 실패하며 반드시 CAPA를 관리하는 사람들의 노력으로 실패하는 것은 아니다. 조직은 종종 CAPA 단계를 완전히 문서화하지 않고 구현 검증과 효과 활동의 검증을 혼동한다.
이 글은 유효성 평가 계획의 CAPA 검증 개발에 사용할 SMART(특정, 측정 가능, 달성 가능, 관련성 및 기한) 방법론을 사용하는 것을 이야기 한다.

Applying The SMART Methodology To CAPAs
유효성 평가에 대한 CAPA 프로세스는 아래 그림과 같이 10 가지 단계로 구성된다. VOEP (verification of effectiveness plan) 8 단계는 거의 사용되지 않지만 유효성 평가 확인 중에 CAPA가 실패 할 수 있다. VOEP는 시정 및 예방 조치가 실제로 효과적인지 확인하기 위해 사전 결정된 기준을 설정하고 문서화하는 데 사용된다.
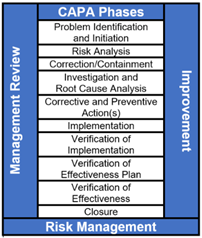
George T. Doran은 1981 년 11 월 미국의 Management Review 호에서 Peter Drucker의 목표 개념 관리를 이용하여 SMART라는 용어를 사용하여 처음으로 인정되었다. 오늘날 SMART 방법론은 일반적으로 개인 목표 및 성과 목표와 관련이 있지만, 방법론은 CAPA VOEP(verification of effectiveness plan) 활동에 쉽게 적용 할 수 있다.

SMART VOEP를 개발하고 평가할 때 다음 질문을 고려해야 한다.
Specific — VOEP가 분명하고 명확하며 집중되어 있는가?
Measurable — 수량화 가능한 데이터가 VOEP를 평가하는 데 사용되는가?
Achievable – VOEP가 실현 가능한가?
Relevant — VOEP가 위험 수준에 적합한가?
Time-Bound — VOEP에 현실적인 마감 시간이 있는가?
Examples of a VOEP’s statements include: VOEP 결과의 예는 다음과 같다.
마지막 3 개의 생산 로트에서 문제가 발생하지 않았다.
지난 3 개월 동안 문제가 다시 발생하지 않았다.
2020 년 2 분기 첫 패스 수율은 90 %에서> 99 %로 증가한다.
스크랩 비율은 3 개월 동안 5 % (예 : 15 %에서 10 %) 감소한다.
유효성 평가 단계는 기준을 검토하고 종결 전에 VOEP에서 사전 확인된 증거를 문서화하는 프로세스이다. 유효성 평가 활동에서 사전 확인된 승인 기준이 충족되지 않으면 CAPA가 실패한다. 유효성 평가에 실패하면 CAPA를 닫고 새로운 CAPA를 Open 하는 것이 적절하다. 또한, 새로운 CAPA가 이전 CAPA와 Tracking이 되는지 확인 할 필요가 있다.

VOEP Example
얼마 전 미국의 어느 제약회사에서 Heat Sealer란 기기를 이용하여 진행되는 패키지 밀봉 공정은 일반적으로 약 1,000개의 제품을 생산하며, 정상적인 근무 시간 내에 모두 완료 되었다. 그러나 때때로 승인된 규격을 충족하지 못하는 제품을 생산하기 시작했다.
생산 책임자는 부적합한 제품 생산의 결과로 CAPA 2020-005를 열었다. CAPA 팀이 구성되었고, 영향 평가가 수행되었으며, 결함이 있는 패키지 밀봉과 관련된 위험으로 인하여 해당 제품은 격리 조치가 되었다. 지난 1년간 생산기록, 일탈, 교정, 유지보수기록, 교육기록, validation 등을 검토하였고, Heat sealer의 엔지니어를 인터뷰하며, 근본 원인 조사를 실시하고, 근본 원인을 파악 한 후 시정 조치를 실시하였다.
생산기록, 일탈, 교정, 유지보수기록, 교육기록, validation 등을 검토하였고, Heat sealer의 엔지니어를 인터뷰를 검토하는 동안 CAPA 팀의 조사에 따르면 Heat Sealer의 장비 설정이 때때로 경험이 부족한 작업자가에 의해 검증된 운전범위 이외로 설정되었다고 결론지었다. CAPA 팀은 이것이 부적합 패키지 밀봉의 근본 원인이라고 결론지었다.
CAPA 팀은 제어시스템의 사용자 액세스 권한을 변경하여 근본 원인을 해결하기로 결정했으며, 이로 인해 작업자는 Heat sealer의 검증 된 운전범위의 설정을 벗어난 운전인자를 선택하는 기능이 효과적으로 제한되었다. Heat sealer 운전자는 검증 된 운전범위 사용의 중요성을 강화하도록 재교육 될 것이다. 또한 CAPA 팀은 실제 Heat sealer 설정을 생산의 시작과 끝에 기록하도록 하여 설정이 검증된 범위 내에 있는지 검증하도록 하였다.
생산 책임자는 CAPA 팀과 함께 SMART 방법을 사용하여 VOEP를 개발했다. CAPA 팀은 다음과 같은 VOEP를 결정했다. Heat Sealer에 의한 패키지는 샘플링 절차에 따라 강화 된 검사 수준을 사용하여 3 일 연속 생산 (3 로트) 동안 규격을 준수하는지 모니터링 한다. CAPA는 향후 3일 연속 생산(3개 로트) 동안 생산된 모든 패키지가 시험 규격을 충족하고, 작업자가 검증된 범위를 벗어난 운전인자를 선택할 수 없으며, 적절한 유효성 검사를 배치 기록에 기록하면 유효하다고 간주된다.
CAPA 팀은 Heat sealer 작업자가 재교육되었는지, 제어시스템에 사용자 접근 권한으로 작업자의 Heat sealer 에 대한 검증된 설정 이외의 운전인자 선택을 제한하고, 생산 기록 문서를 문서화하여 문서화했는지 검증하였다. CAPA 팀은 실행 과제에 대한 검증이 계획대로 완료되었다고 결론지었다.
다음 단계에서 CAPA 팀은 VOEP를 평가하여 SMART인지 여부를 판단했다.
Specific — VOEP가 분명하고 명확하며 집중되어 있습니까? ☑ 예 ☐ 아니오
Heat Sealer에서 3 일간 연속 생산 (3 Lot) 동안 생산 된 패키지는 풀 테스트 규격을 충족해야 한다.
작업자는 검증 된 범위를 벗어난 운전인자를 선택할 수 없다.
적절한 유효성 평가 설정이 배치 기록에 기록된다.
Measurable — 수량화 가능한 데이터가 VOEP를 평가하는 데 사용됩니까? ☑ 예 ☐ 아니오
Heat Sealer에서 3 일간 연속 생산 (3 Lot) 동안 생산 된 패키지는 풀 테스트 규격을 충족해야 한다.
작업자는 검증 된 범위를 벗어난 운전인자를 선택할 수 없다.
적절한 유효성 평가 설정이 배치 기록에 기록된다.
Achievable — VOE가 실현 가능하거나 실용적입니까? ☑ 예 ☐ 아니오
Heat Sealer에서 3 일간 연속 생산 (3 Lot) 동안 생산 된 패키지는 풀 테스트 규격을 충족해야 한다.
Relevant — VOE가 위험 수준에 적합한가? ☑ 예 ☐ 아니오
Heat Sealer에 패키지는 샘플링 절차에 따라 강화 된 검사 수준을 사용하여 3 일 연속 생산 (3 Lot) 동안 규격을 준수하는지 모니터링 한다.
Time-Bound — VOE에 마감 시한이 있습니까? ☑ 예 ☐ 아니오
연속 3 일 동안 생산 된 패키지 (3 Lot)
CAPA 팀은 다음과 같은 결과를 통해 VOE를 수행했다.
확인 된 설정을 사용하여 3 일 연속 생산 (3 lot) 동안 Heat Sealer 을 가동했다. ☑ 예 ☐ 아니오
모든 패키지가 풀 테스트 규격을 충족한다. ☑ 예 ☐ 아니오
각 생산 작업의 시작과 끝에 대해 검증 된 Heat sealer 설정이 배치 레코드에 올바르게 기록되었다. ☑ 예 ☐ 아니오
CAPA 팀은 CAPA가 완전하고 효과적이라고 결정했다. 또한 팀은 다른 부서와 성공을 공유하여 유사한 조치를 수행 할 수 있었다.
이 예에서는 CAPA 효과를 계획하고 확인하기 위해 SMART 방법을 적절히 사용하여 CAPA를 closing하는 방법을 보여준다. SMART 방법을 적용하면 CAPA 프로그램의 효율성을 높이고 규정, 표준 및 기대치 준수를 입증 할 수 있다.

Conclusion
규정 및 지침에 따라 CAPA가 요구 될 뿐만 아니라 CAPA는 올바르게 수행 될 경우 폐기를 줄이고 프로세스를 개선하여 조직의 경쟁력을 향상시킬 수 있다. SMART 방법론을 사용하면 유효성 평가 계획을 개발하여 CAPA 시스템을 준수 할 수 있다.
다음은 CAPA와 관련된 몇 가지 규정을 소개하며, 오역을 예방하기 위하여 원문을 기술한다.
21 CFR 820 Quality System Regulations
Sec. 820.100 Corrective and preventive action:
(a) Each manufacturer shall establish and maintain procedures for implementing corrective and preventive action. The procedures shall include requirements for:
Analyzing processes, work operations, concessions, quality audit reports, quality records, service records, complaints, returned product, and other sources of quality data to identify existing and potential causes of nonconforming product, or other quality problems. Appropriate statistical methodology shall be employed where necessary to detect recurring quality problems;
Investigating the cause of nonconformities relating to product, processes, and the quality system;
Identifying the action(s) needed to correct and prevent recurrence of nonconforming product and other quality problems;
Verifying or validating the corrective and preventive action to ensure that such action is effective and does not adversely affect the finished device;
Implementing and recording changes in methods and procedures needed to correct and prevent identified quality problems;
Ensuring that information related to quality problems or nonconforming product is disseminated to those directly responsible for assuring the quality of such product or the prevention of such problems; and
Submitting relevant information on identified quality problems, as well as corrective and preventive actions, for management review.
(b) All activities required under this section, and their results, shall be documented.

ISO 13485:2016 Medical devices — Quality management systems — Requirements for regulatory purposes
8.5.2 Corrective action
The organization shall take action to eliminate the cause of nonconformities in order to prevent recurrence. Any necessary corrective actions shall be taken without undue delay. Corrective actions shall be proportionate to the effects of the nonconformities encountered.
The organization shall document a procedure to define requirements for:
a) reviewing nonconformities (including complaints);
b) determining the causes of nonconformities;
c) evaluating the need for action to ensure that nonconformities do not recur;
d) planning and documenting action needed and implementing such action, including, as appropriate, updating documentation;
e) verifying that the corrective action does not adversely affect the ability to meet applicable regulatory requirements or the safety and performance of the medical device;
f) reviewing the effectiveness of corrective action taken.
Records of the results of any investigation and of action taken shall be maintained.
8.5.3 Preventive action
The organization shall determine action to eliminate the causes of potential nonconformities in order to prevent their occurrence. Preventive actions shall be proportionate to the effects of the potential problems.
The organization shall document a procedure to describe requirements for:
a) determining potential nonconformities and their causes;
b) evaluating the need for action to prevent occurrence of nonconformities;
c) planning and documenting action needed and implementing such action, including, as appropriate, updating documentation;
d) verifying that the action does not adversely affect the ability to meet applicable regulatory requirements or the safety and performance of the medical device;
e) reviewing the effectiveness of the preventive action taken, as appropriate. Records of the results of any investigations and of action taken shall be maintained.

The Joint IPEC - PQG Good Manufacturing Practices Guide for Pharmaceutical Excipients.
8.5.2 Corrective Action
The excipient manufacturer should establish, document and maintain procedures for:
l determining the root causes of nonconformities,
l ensuring that corrective actions are implemented and effective,
l Implementing and recording changes in procedures resulting from corrective action.
8.5.3 Preventive Action
The excipient manufacturer should establish, document and maintain procedures for:
l initiating preventive actions to deal with problems at a level corresponding to the risks,
l Implementing and recording changes in procedures resulting from preventive action.

ICH Harmonised Tripartite Guideline Pharmaceutical Quality System Q10
3.2.2 Corrective Action and Preventive Action (CAPA) System
The pharmaceutical company should have a system for implementing corrective actions and preventive actions resulting from the investigation of complaints, product rejections, non-conformances, recalls, deviations, audits, regulatory inspections and findings, and trends from process performance and product quality monitoring. A structured approach to the investigation process should be used with the objective of determining the root cause. The level of effort, formality, and documentation of the investigation should be commensurate with the level of risk, in line with ICH Q9. CAPA methodology should result in product and process improvements and enhanced product and process understanding.

'My Job > Pharmaceutical Quality System' 카테고리의 다른 글
| Zero-Particulates 목표는 지속적인 개선을 촉진한다. (0) | 2022.10.31 |
|---|---|
| Change Control Procedure in Pharmaceuticals (변경관리) (0) | 2022.10.31 |
| 제약의 Risk management를 위한 10 가지 실용적인 팁 (0) | 2022.10.26 |
| 문제의 근본 찾기 (Root Cause Analysis–Finding the Root of the Problem) (0) | 2022.10.18 |
| The Product Quality Leader(PQL) Team Part I: PQL의 역할 구축 (0) | 2022.10.18 |