바이오 의약품의 복잡성과 프로세스가 계속 증가함에 따라 개발 및 제조를 지원하는 데 필요한 품질 관리 시스템 (QMS, Quality Management System)은 설계 및 구현이 더욱 어려워지고 있습니다. QMS는 제조 작업이 다양한 적용 가능한 규정 및 지침 (R&G, regulations and guidelines)을 준수하도록 유지하면서 모든 위험을 허용 가능한 수준으로 완화하기 위해 광범위한 제어 전략 (CS, control strategies)이 필요합니다. QMS는 건강, 안전 및 환경 (HSE, health, safety, and environmental) 위험, 요구 사항 및 규정을 포함해야 하기 때문에 여기에서 사용 된 QMS는 PQS(product quality system)의 상위 집합입니다. 예를 들어, 일부 제품의 직원 안전은 CS 개발에서 환자 안전 및 효능만큼 중요 할 수 있습니다. 이 기사에서는 21 세기에 효율적이고 효과적이며 호환되는 QMS를 구축하기 위해 QMS를 개발하고 구현하는 방식의 근본적인 변화를 제안합니다.

QMS는 다음의 세 가지 목표 또는 설계 기준을 충족해야 하는 상호 연결된 CS의 집합이다
1. 제어 제공 – 모든 위험을 허용 가능한 수준으로 최소화하고 CS가 모든 프로세스의 설계 기준을 충족하기 위해 필요한 수행을 보장하기 위해 프로세스 또는 활동에 대한 통제력을 확립하고 유지한다.
2. 통제 증명 – 회의적인 외부집단에 통제목표가 충족되었다는 문서화된 증거를 제공한다.
3. 통제력 향상 – 제어 또는 제어 증명이 충족되지 않으면 프로세스 또는 시스템을 제어 상태로 신속하게 되돌릴 수 있는 메커니즘을 제공하고 향후 제어가 유지 될 수 있도록 CS를 개선해야 한다.
세 가지 CS 목표를 항상 동시에 달성해야 한다는 요구 사항으로 인해 바이오 의약품 제조는 대부분의 다른 제조 작업과 분리된다. 통합 QMS는 세 가지 목표, 특히 세 번째 목표를 동시에 달성하도록 설계되어야 한다. 통제 개선의 목표는 근본 원인 분석 (RCA, root cause analysis) 제어 전략과 수정 및 예방 조치 (CAPA, corrective and preventive action)를 다른 모든 CS에 통합해야 한다. 지속적인 개선을 위한 RCA 및 CAPA는 제어 목표 또는 실현 된 위험 결과와의 편차뿐만 아니라 거의 누락 된 부분을 식별하고 수정해야 한다.
QMS 개발을 위한 보편적이거나 기존의 방법은 비교적 조절 및 제어가 쉬운 매우 간단한 화학 제품으로 시작하여 수십 년 동안 발전해 왔다. 제품과 그 중간체는 제조 결함에 대해 테스트 될 수 있기 때문에, 초기 CS 접근법은 주로 중간체와 제품의 문서화 된 테스트를 통해 제어를 제공하는 첫 두 가지 목표를 달성하는 것으로 구성되었다. 제품과 프로세스가 복잡 해짐에 따라 필요한 제조 관행을 간단하고 적절하고 충분한 표준으로 명시하는 것이 점점 어려워지고 있다.
Conventional QMS Design Approach
일반적인 접근 방식은 규정을 검토하고 이해함으로써 규정 준수를 달성하는 것이다. 규제는 매우 모호하고 다양한 상황, 제품 및 프로세스에 적용되기 때문에 다양한 해석을 요구한다. 규정은 기본적으로 충분하고 적절한 기준에 따라 허용 가능한 최소 벤치 마크를 설정하는 데 목적이 있으며 설계 표준으로 사용되지 않는다. 복잡한 생물학적 제제에 대해 수용 가능한 최소 표준을 정의하는 것은 매우 어렵다. 경우에 따라 불확실성이 높고 알려지지 않은 요인으로 인해 허용 가능한 최소 표준으로 간주 될 수 있는 수준을 훨씬 뛰어 넘는 것이 적절할 수 있다. 더욱 포괄적인 표준에 대한 요구가 증가함에 따라 규정을 해석하기가 어려워짐에 따라 규제 기관이 QMS 개발을 위한 설계 기준으로 규정을 사용하는 데 도움을 주기 위해 여러 가지 지침을 마련하기 시작했다. 그러나 규제 기관은 지나치게 규범적인 것을 원하지 않고 특정 상황에 대한 과도한 복잡성 및 잘못된 적용을 초래할 수 있으므로 지침도 다양한 설정과 상황에 유연하게 적용된다.
대형 생물학적 분자부터 첨단 치료용 의약품 (ATMP, advanced therapeutic medicinal products)에 이르기까지 광범위한 유형의 제품 및 치료제와 복합적으로 사용 될 때 R&G의 수와 특성은 상호 작용, 중복, 일치하지 않는 문서가 폭발적으로 증가함에 따라 이를 완전히 포괄적으로 읽고, 검토하고, 이해하는 것은 거의 불가능하다. 예를 들어, GTP(good tissue practices)는 거의 동일한 개념, 접근 방식 및 요구 사항을 가진 GMP와 매우 유사하다. 조직을 취급하고 처리하는 것이 제품을 제조하는 것으로 간주되는 경우 GTP와 GMP는 매우 유사한 위험을 제어하므로 사실상 동일한 목표를 가지고 있다.
QMS 개발을 위해 무수한 R&G를 사용하는 현재의 접근 방식이 그림 1에 요약되어 있다. 복잡한 제품 및 프로세스를 고려할 때 개발 및 제품의 Life cycle 동안 특정 프로세스 및 제품에 적용될 수 있는 모든 R&G를 이해하는 것은 매우 어려울 수 있다.
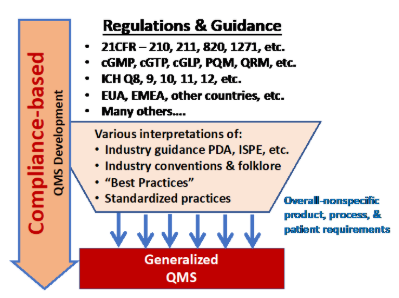
Figure 1: QMS를 개발하는 기존의 방법은 정부 규제 및 지침 (R&G)에서 필요한 제어 전략을 도출하는 것이다. 많은 R&G는 개발 및 제조를 제어하도록 설계된 QMS에 적용하기 위한 다양한 해석 및 관점을 통해 처리되며, 그 결과 QMS는 일반적으로 다양한 상황과 위험을 제어하도록 설계된 일반화 된 시스템이다.
많은 사람들이 문제를 해결하기 위해 적용 가능한 모든 R&G를 읽고 연구하지 않는다. 따라서 많은 QMS는 불완전한 규제 표준을 기반으로 하기 떄문에 결과적으로 많은 QMS 설계는 회사가 과거에 다른 제품 및 프로세스에 대해 본 내용을 기반으로 하였으며, 많은 회사들이 다양한 제품, 프로세스 및 제조 시설에서 균일 성을 달성하기 위해 QMS를 표준화하려고 시도했다.
FDA의 2006 Guidance for Industry: Quality Systems Approach to Pharmaceutical CGMP Regulation의 3 페이지에는 다음 내용이 포함된다.
A quality system adopted by a manufacturer can be tailored to fit the specific environment, taking into account factors such as scope of operations, complexity of processes, and appropriate use of finite resources.
물론 핵심 문제는 운영 범위와 프로세스의 복잡성에 따라 유한 자원을 적절히 사용하도록 QMS를 조정하는 방법이다.
그림 1과 같이 매우 많은 수의 복잡한 R&G를 사용하여 QMS를 구축하기 위한 기본 설계 접근 방식이 특정 제품, 프로세스 및 시설 위험만 제어하도록 설계된 접근 방식으로 변경하는 경우보다 효율적이고 효과적인 QMS를 구축 할 수 있을까?
The Risk-Based QMS Design Approach
대안은 개발, 제조 과정에서 제품, 제조 공정, 직원 및 환자에게 노출 될 특정 위험을 식별하고 평가하여 QMS를 구축하는 것이다. 그림 2에 나와 있는 기본 도구를 사용하여 아래에 간략하게 설명된 위험을 식별 할 수 있을 것이다.
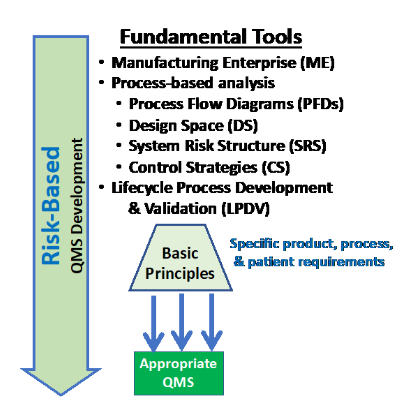
Figure 2: 위험 관리 방식을 사용하여 제조 기업 (ME, manufacturing enterprise)의 일부로 필요한 제어 전략을 정의하는 데 중점을 둔 기본 도구와 패러다임을 사용하여 적절한 QMS를 개발할 수 있다.
QMS는 모든 ME의 위험을 통제하기 위해 함께 작동하는 다수의 제어 전략 또는 제어 시스템을 종합 한 것이다. 합리적으로 발생할 수 있는 ME 및 제품에 대한 모든 위험을 식별 및 관리하고 이러한 위험을 제어하는 데 필요한 CS를 구축하기 위해 4 가지 도구 또는 접근 방식을 사용할 수 있다.
4 Fundamental Tools
네 가지 기본 도구는 ME가 어떻게 구성되어 있는지 이해하는 것으로 시작 한다. 프로세스 기반 분석을 사용하여 제품을 제조하는 데 필요한 모든 프로세스를 식별함으로써 모든 프로세스와 관련된 위험을 시스템 위험 구조 (SRS, system risk structures)를 사용하여 식별, 평가, 분석 및 완화 할 수 있다. LPDV (Life Cycle Process Development and Validation) 도구를 사용하여 CS 프로세스를 개발 또는 개선하여 수용 할 수 없는 위험을 제어하거나 완화 할 수 있다.
1. Manufacturing Enterprise
공정 개발에서 상업용 제조에 이르는 모든 제품 제조는 제조 공정, 시설 및 인프라의 세 가지 요소로 구성된 기업에서 나온다.
* 제조 공정: 제품 제조에 관련된 작업 (UO, unit operations), 장비, 계측기, 배관 및 원자재로 제품 생산을 직접 처리하거나 영향을 미치는 모든 시스템.
* 시설: 자재, 인력, 장비 등의 흐름과 제조 공정을 둘러싼 기능을 포함한 회의실 레이아웃. WFI와 같은 원자재를 공급하는 중앙 또는 공용 유틸리티 시스템도 포함.
* 인프라 (QMS): 제조 프로세스 및 시설 요소의 운영을 제어하는 운영 및 제조 절차, 실무, 정책 및 기타 지원 시스템.
요약하면, ME는 인프라의 제어 하에 설비에 의해 포함되고 지원되는 제조 프로세스로서 기술 될 수 있다.
2. Process-based analysis
세 가지 ME 요소는 모두 프로세스로 세분 될 수 있다. 이러한 프로세스는 제품 제작의 모든 측면을 제어하는 상호 의존적 인 시스템의 네트워크를 형성한다. 이러한 프로세스는 프로세스 플로우 다이어그램 (PFD, process flow diagrams)을 사용하여 연결 및 프로세스 간의 관계와 상호작용을 매핑 할 수 있다. 단위 작업 네트워크로 설명 된 제조 프로세스의 경우 모든 프로세스 입력을 UO의 위협을 식별하는 작동 매개 변수, 입력 재료 속성 또는 장비 매개 변수 범주에 배치하는 각 UO의 설계 공간 (DS)을 개발할 수 있다.
3. System Risk Structures (SRS)
PFD는 프로세스에 대한 입력 위협이 다양한 위험 결과를 초래할 수 있는 방법을 설명하기 위해 필요한 SRS를 구축하기 위한 기초로 사용될 수 있다. 그런 다음 전문가 팀이 SRS를 분석하여 PCRM(prospective causal risk modeling)을 사용하여 위험 결과가 발생할 가능성을 확인한다. SME(Subject Matter Expert)는 위험을 적절하게 통제할 수 없기 때문에 프로세스를 통과하는 위험의 가능성과 결합하여 실제 위험이 발생할 가능성을 분석한다. 그 후 CS 프로세스를 변경하거나 추가하여 발생할 수 없는 위험을 통제하거나 완화한다.
4. Life Cycle Process Development & Validation (LPDV)
모든 공정은 ME의 제조 공정, 시설 또는 인프라 요소의 일부에 관계없이 네 가지 기본적인 질문을 가지고 있다:
* 프로세스 설계의 목표는. – 정의(stage #0)
* 이 과정은 어떻게 목표를 달성할 것 인가. – 설계 (stage #1)
* 프로세스 설계가 잘될까. – 자격(stage #2)
* 프로세스 실행은 제대로 이뤄졌나. – 운영 및 확인(stage #3)
4 가지 질문은 SOP, 단위 작업, 장비, 클린룸 작업, 교육, 레이아웃을 통한 원자재 흐름 등 모든 프로세스에 공통적이다. 가장 중요한 단계는 다음 두 실행 단계에서 디자인 단계 (stage # 1)이다. QbD (Quality by Design)를 사용하여 질문에 답변함으로써 설계 단계부터 거의 완전히 개발되고 검증 된 프로세스를 제공한다. 4 단계 LPDV 패러다임은 FDA의 혁신적인 프로세스 검증 지침의 진화다.
위에서 설명한 네 가지 도구는 다양한 위험을 제어하고 수용하는 데 필요한 CS를 구축하는 기초로 다양한 위험을 식별, 처리 및 문서화하기 위한 비교적 간단한 순차적 패러다임을 제공한다.
The All-Important Final Step
모든 QMS 요소는 모든 규정을 준수해야 한다. 마지막 단계는 다양한 R&G에 대해 위험 기반 QMS를 검토하여 규제 요구 사항을 충족하고 적절한 지침을 준수하는지 확인하는 것이다. 모든 다양한 QMS 요소에 대한 규정 및 지침은 종종 중복되고 모호하며 다소 혼란스럽지만, 포괄적이고 효과적인 QMS를 구축하기 위한 많은 지혜와 중요한 고려 사항이 포함되어 있으며, 적절한 규제 문서에 대한 철저한 지식이 필수적이며 정보를 뒷받침하는 데 유용 할 수 있다. 기본 도구와 기본 위험 완화 및 제어 개념을 사용하여 QMS를 설계 한 경우 규제 문서를 기준으로 해당 요소를 확인하는 것이 훨씬 쉬워지며, SRS로 돌아가서 CS 프로세스를 적절히 추가 또는 수정하여 필요한 제어 및 준수를 제공함으로써 차이를 식별하고 해결할 수 있다.

Conclusion
그림 1에 나와있는 현재 QMS 개발 접근 방식이 얼마나 강력한지를 감안할 때 그림 2에 나와있는 보다 기본적인 접근 방식으로 전환하기는 어렵다. 프로세스 흐름 다이어그램, 디자인 공간 및 시스템 위험 구조를 구성하고 검토 할 때 디자인 프로세스 전체에서 소규모로 시작하여 시스템을 구축하는 것이 좋다. 설계하는 동안, ME 요소들 간의 상호 작용은 상호 작용 위험에 대한 필요한 제어를 완료하기 위해 추가적인 CS를 개발하고 향상시켜야 할 것이다. 새로운 방법은 처음에는 시간과 노력을 절약 할 수는 없지만 더 나은 QMS를 생성하고 결국에는 시간과 노력을 절약 할 수 있기 때문에 이 접근법이 권장된다.
그러나 주요 이점은 방법을 사용하지 않고 남겨둔 문서에서 비롯됩니다. SRS는 적절한 위험 기록 및 요약과 결합 될 경우 내부 회사 검토 및 합의 및 규제 기관에 대한 QRM의 설계에 대한 효과적인 문서를 제공한다. SRS를 사용하면 CS를 통해 식별 된 위협의 흐름을 효과적으로 설명하여 위험 결과의 개선을 보여줄 수 있다. 또한 LPDV를 사용하면 특히 단계 # 1 설계 활동 중에 QbD 연습을 사용하는 경우 PDF 및 SRS에 제어 프로세스를 수정하거나 추가하여 CS를 개발하고 구현 한 방법에 대한 추가 문서가 제공된다. 컴플라이언스 기반 QMS 설계의 주요 문제점 중 하나는 다양한 R&G를 인용하는 것 이외의 다양한 위험을 제어하기 위해 사물을 설계 한 이유 또는 방법에 대한 특정 문서가 사실상 없다는 것이다.